NDSRIs (Nitrosamine Drug Substance–Related Impurities): Regulatory Risk, Analytical Challenges, and Control Strategies
Nitrosamine Drug Substance–Related Impurities (NDSRIs) have emerged as a critical concern for pharmaceutical manufacturers and regulators worldwide. Unlike simple nitrosamines, NDSRIs are structurally related to the active pharmaceutical ingredient (API) and may form during synthesis, processing, or storage. Because many nitrosamines are classified as probable human carcinogens, regulatory agencies require robust risk assessment, sensitive analytical detection, and effective control strategies.
As regulatory scrutiny increases, pharmaceutical companies must adopt science-based approaches to identify, quantify, and mitigate NDSRIs to ensure patient safety and product compliance.
What Are NDSRIs?
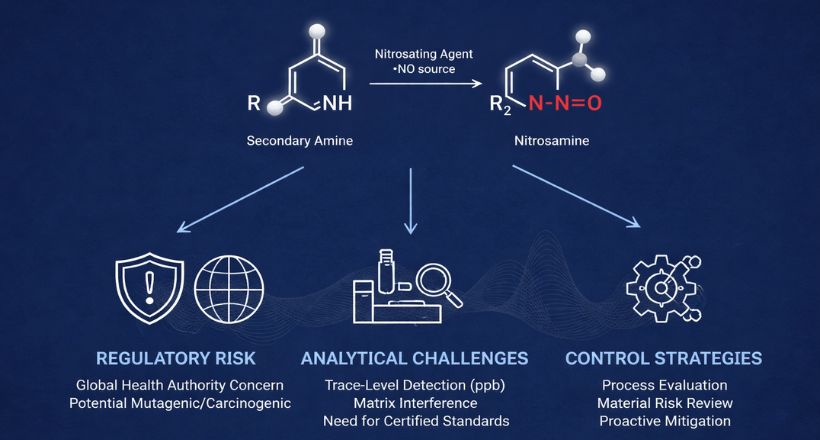
NDSRIs are nitrosamine impurities formed from amine-containing drug substances or intermediates. They typically arise when secondary or tertiary amines react with nitrosating agents such as nitrites under favorable conditions. These impurities are particularly concerning because:- they may be structurally similar to the API
- they can form under routine manufacturing conditions
- they often occur at trace levels requiring ultra-sensitive detection
Why Regulators Are Concerned
Global health authorities, including the U.S. Food and Drug Administration, European Medicines Agency, and World Health Organization, require manufacturers to evaluate nitrosamine risks due to their potential mutagenic and carcinogenic properties.
Key regulatory expectations include:
- risk assessment for nitrosamine formation pathways
- establishment of acceptable intake (AI) limits
- validated analytical methods capable of detecting trace levels
- implementation of mitigation and control strategies
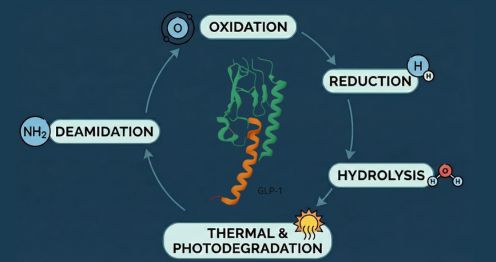
Formation Pathways of NDSRIs
Understanding formation mechanisms is essential for effective risk mitigation. NDSRIs may form through:- Nitrosation Reactions: Secondary or tertiary amines react with nitrosating agents (e.g., nitrites) under acidic or process-specific conditions.
- Raw Material and Reagent Impurities: Nitrite contamination in solvents, reagents, or excipients can trigger nitrosamine formation.
- Process Conditions: Factors influencing formation include:
- low pH environments
- elevated temperatures
- prolonged hold times
- presence of oxidizing agents
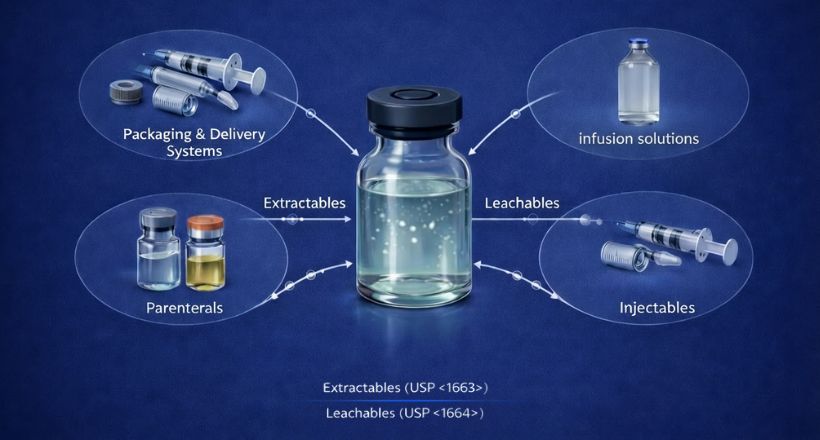
- Storage and Packaging Interactions: Nitrosamines may form during storage due to degradation pathways or interactions with packaging components.
Analytical Challenges in Detecting NDSRIs
Detecting NDSRIs presents significant analytical complexity due to their structural diversity and trace-level presence.Key challenges include:
- detection at parts-per-billion (ppb) or lower levels
- matrix interference from complex drug formulations
- need of reference standards for NDSRIs
- need for structural confirmation
Advanced analytical approaches
Highly sensitive techniques such as LC-MS/MS are commonly employed for screening and quantification. HRMS supports structural elucidation of unknown impurities.Risk Assessment and Control Strategies
A proactive, science-based approach is essential to control NDSRIs.Risk assessment steps:
- Process evaluation Identify amine sources and potential nitrosating agents.
- Material risk review Assess nitrite contamination risks in raw materials and excipients.
- Laboratory stress studies Simulate process conditions to evaluate nitrosamine formation potential.
- control nitrite levels in materials and water systems
- adjust process pH and temperature conditions
- minimize hold times during manufacturing
- incorporate antioxidants or inhibitors where appropriate
- implement protective packaging strategies
Importance of Certified Standards and Method Validation
Accurate quantification and regulatory compliance depend on reliable certified standards and fully validated analytical methods. High-quality impurity standards support:- method development and validation
- structural confirmation
- regulatory submissions
- routine quality control
How Daicel Chiral Technologies India Supports NDSRI Analysis
Daicel Chiral Technologies India provides advanced analytical solutions to support pharmaceutical companies in addressing nitrosamine and NDSRI challenges.Capabilities include:
- development of sensitive LC-MS /MS methods for NDSRI detection
- impurity identification and structural characterization
- large inventory of high-purity certified NDSRI standards
- stable isotope labelled NDSRI standards to mitigate matrix affects
- risk assessment support aligned with global regulatory expectations
- method validation and batch testing for regulatory compliance