Extractables & Leachables: The Silent Variables in Drug Safety
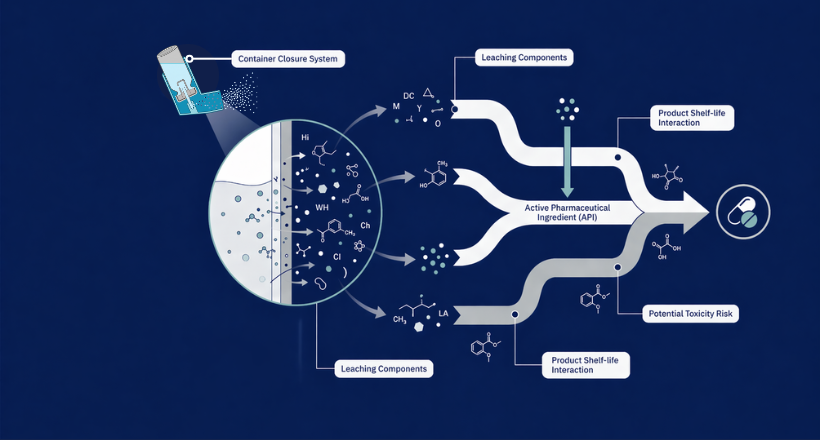
There is a question that rarely makes it onto a product specification sheet but sits at the center of every drug approval conversation: what does the container do to the drug? Not what it prevents — microbial ingress, oxidation, moisture — but what it quietly introduces. The chemicals that migrate from a polymer cap, a rubber stopper, a silicone-coated syringe, or an infusion bag into the formulation itself.
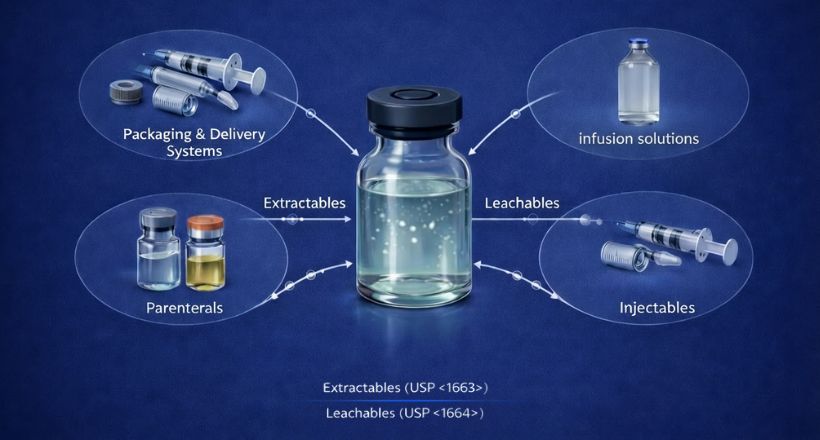
This is the domain of Extractables and Leachables — a field that has moved from regulatory footnote to front-of-line concern as biologics, inhalation products, and prefilled delivery systems have become routine. The science is deceptively simple. The regulatory machinery around it is not.
Defining the Terms
Extractables are chemical compounds that can be released from a container-closure system or drug delivery component under controlled laboratory conditions — typically using aggressive solvents, elevated temperatures, and extended contact times. Think of it as a worst-case simulation: what is the maximum the material could ever give up?
Leachables: by contrast, are compounds that actually migrate into a drug product under normal conditions of storage and use. They are a subset of extractables, filtered through the real-world chemistry of the formulation — its pH, polarity, storage temperature, and time on shelf. Where extractables define the universe of potential concern, leachables define what the patient is actually exposed to.
"Extractables characterize the container. Leachables characterize the drug product. The gap between them is where risk assessment lives."
Why It Matters More Now
The pharmaceutical landscape has shifted considerably. Complex biologics delivered through wearable injectors. Inhalation therapies with fine-particle aerosols passing directly through the lungs. Prefilled syringes with elastomeric components in sustained contact with protein-based formulations. Each of these formats creates intimacy between material and medicine that solid oral dosage forms simply do not. Regulators have responded. In August 2025, ICH advanced the long-anticipated Q3E guideline to Step 2b — the start of the global public consultation phase. This represents a decisive shift toward globally harmonized, science-based E&L requirements, addressing a regulatory gap that has historically created variability across markets. The expectation is not just that you test — it is that you test systematically, connect the data to toxicological thresholds, and demonstrate understanding rather than just compliance.The Regulatory Framework
Several overlapping standards govern how E&L studies are designed, executed, and interpreted. These are not entirely harmonized, which creates its own challenge for organizations working across markets simultaneously. Separation is influenced by:- USP <1663> / <1664> — Assessment of extractables from packaging systems and leachables in drug products. Provides risk-tiered testing guidance and is the most commonly cited reference for US submissions.
- ICH Q3E — The emerging harmonized guideline addressing elemental and organic extractables and leachables. Expected to align global practice significantly once finalized.
- ISO 10993 Series — Biological evaluation of medical devices. Extensively cross-referenced in combination product E&L programs, particularly for drug-device delivery systems.
- PQRI Recommendations — Industry guidance for orally inhaled and nasal drug products — one of the most operationally detailed references available, widely adopted even beyond its original scope.
The Testing Workflow
An E&L program is not a single study — it is a sequence of decisions, each one narrowing the field of concern. Understanding the logic of that sequence matters as much as the analytical methods themselves.- Phase 1 — Risk assessment & material qualification — Establishing what components are in contact with the formulation, for how long, and with what surface area. Material data sheets and supplier documentation anchor this phase.
- Phase 2 — Extractables study design — Selecting solvents, temperatures, and extraction conditions that stress the material appropriately. ICH and USP frameworks provide the structure; scientific judgment calibrates the design to route and clinical risk.
- Phase 3 — Analytical characterization — Typically involves a combination of GC-HS-MS/MS, HRMS, ICP-MS and IC. The objective is to achieve broad, untargeted screening coverage followed by targeted identification and confirmation of detected compounds.
- Phase 4 — Threshold-based filtering — Applying the AET, Qualification Threshold (QT), and Safety Concern Threshold (SCT) to determine which detected compounds require further characterization.
- Phase 5 — Leachables correlation — Comparing real-world leachables data from stability samples against the extractables profile to confirm that actual migration aligns with predictions.
- Phase 6 — Toxicological risk assessment — Reviewing each compound of concern against established safety thresholds using literature precedent, TTC, or compound-specific safety data.
Key Thresholds and Their Logic
The Analytical Evaluation Threshold (AET) sets the minimum reporting level — compounds detected below it may be acknowledged but do not require further action. The Safety Concern Threshold (SCT), most clearly articulated in the PQRI recommendations for orally inhaled products, represents the daily intake level below which a leachable is unlikely to pose safety concern regardless of its chemical nature. Between these sits the qualification zone, where compound identity, structure, and toxicological precedent all come into play. Threshold values vary meaningfully by route of administration. A compound acceptable at a given level for an oral product may not clear the bar for a pulmonary or ophthalmic formulation, where the protective barriers are different and the patient population may include those with underlying vulnerabilities.Common Leachables of Concern
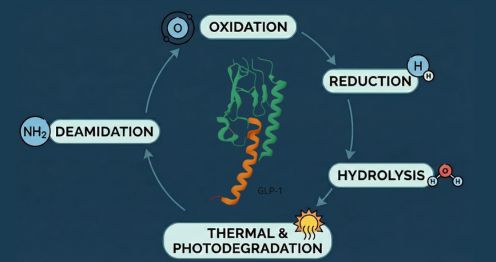
Experience across thousands of E&L programs has identified a recurring cast of chemical migrants. Knowing them shapes how studies are designed and where analytical attention is concentrated. N-nitrosamines · Antioxidants (BHT, BHA) · Plasticizers (DEHP, DINP) · Photoinitiators · 2-Mercaptobenzothiazole · Rubber oligomers · Elemental impurities (Ni, Cr, Ti, Co) · Formaldehyde / Acetaldehyde · Processing aids & lubricants N-nitrosamines deserve particular attention. They have attracted intense regulatory scrutiny since their detection in sartan and ranitidine products, and current expectations from both FDA and EMA require proactive risk evaluation. The intersection of nitrosamine risk management and traditional E&L frameworks is an area where regulatory guidance continues to evolve rapidly.How Daicel Addresses E&L Challenges — End to End
One of the structural difficulties in E&L work is that it demands capability across two very different domains simultaneously: the analytical rigor of a contract research organization and the reference material precision of a standards supplier. Most pharma and medical device companies have to coordinate between these two. Daicel is one of the few organizations that brings both under the same roof.Daicel E&L Study Packages — Purpose-Built for Pharma and Medical Devices
Daicel offers comprehensive E&L studies for a broad range of packaging materials and portfolio products, with a team of scientists committed to providing solutions that meet the specific needs of pharmaceutical companies. The study offering is built around a risk-based approach that keeps the regulatory end goal in focus from day one. Daicel's scientists bring deep experience across formulation types — including the "4I's" (inhalants, injectables, infusions, and implantables), OINDPs, parenterals, peptides and biologics, single-use systems, medical devices, and label/ink migration. Errors in study design at the extractables stage propagate expensively into leachables work and toxicological assessment further downstream.Key capabilities include:
- Regulatory-aligned study design — Daicel's team plans E&L studies in accordance with current regulatory guidelines including USP <1663>, <1664>, PQRI guidance, ISO 10993-18, and the evolving ICH Q3E framework.
- Comprehensive instrumentation — Studies are conducted on GC-HS-MS/MS, LC-MS/MS, High Resolution Mass Spectrometry (HRMS), ICP-MS, and Ion Chromatography (IC) — covering volatile, semi-volatile, non-volatile, and elemental fractions in a single integrated program.
- In-house impurity synthesis — Daicel has a state-of-the-art facility for the isolation and synthesis of impurities observed above AET in leachable studies. When a compound requires a certified E&L standard for method validation, Daicel can synthesize it internally — eliminating the third-party delays that commonly stall E&L programs.
- Validated simulation and leachables methods — Daicel offers validated methods for both simulation studies and leachables studies, providing the methodological foundation required for GMP-compliant stability testing and regulatory submission.
- Project management infrastructure — Communication and reporting are managed through a structured system aligned to customer-specific requirements and timelines, giving all stakeholders synchronized visibility throughout the program.