Analytical Techniques for Oligonucleotide Impurity Analysis and Separation
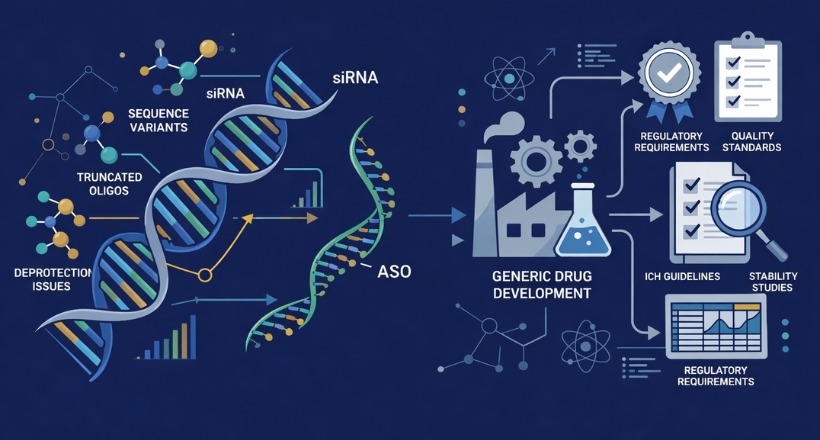
The rapid advancement of oligonucleotide therapeutics has introduced new analytical challenges for pharmaceutical scientists. These nucleic acid–based drugs, including antisense oligonucleotides and siRNA, often contain closely related impurity species that differ from the target molecule by only a single nucleotide or minor chemical modification.
Accurate identification and quantification of these impurities are essential to ensure product quality, safety, and regulatory compliance. However, traditional analytical approaches used for small-molecule drugs are often insufficient for these larger, highly charged biomolecules. As a result, specialized analytical techniques are required.
Chromatographic separation methods, combined with advanced detection technologies, play a central role in oligonucleotide impurity analysis.
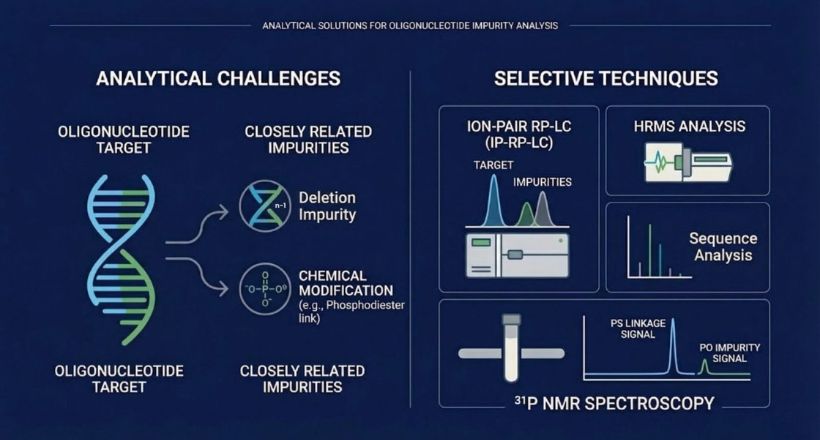
Analytical Challenges in Oligonucleotide Impurity Profiling
Oligonucleotides present several unique analytical challenges:
- High structural similarity: Impurities such as n–1 or n–x truncations, deletion sequences, and mismatch variants differ from the target by only a single nucleotide, making separation difficult.
- Synthesis-related impurities: Solid-phase synthesis can introduce failure sequences, depurination products, and incomplete deprotection species.
- Chemical modifications: Therapeutic oligonucleotides often include modifications such as phosphorothioate linkages or 2′-O-methyl groups, increasing molecular heterogeneity.
- Charge and size effects: The high negative charge density and relatively large molecular size influence chromatographic behavior through counterion interactions and potential secondary structures.
Ion-Pair Reversed-Phase Liquid Chromatography (IP-RP-LC)
Ion-pair reversed-phase liquid chromatography (IP-RP-LC) is one of the most widely used techniques for oligonucleotide analysis. In this method, ion-pairing reagents interact with the negatively charged phosphate backbone of oligonucleotides, enabling their retention on hydrophobic stationary phases. Separation is driven by a combination of ion-pair interactions and apparent hydrophobicity differences. Common ion-pairing systems include:- Triethylammonium acetate (TEAA)
- Hexafluoroisopropanol (HFIP) with triethylamine (TEA), especially for LC–MS applications
Ion-Exchange Chromatography
Ion-exchange chromatography separates oligonucleotides based on their charge interactions with the stationary phase. Since oligonucleotides carry multiple negative charges along their backbone, this technique is well suited for separating sequence variants, truncated species, and impurities differing in charge density. Separation is influenced by:- Charge density
- Sequence length
- Base composition
High-Resolution Mass Spectrometry (HRMS)
High-resolution mass spectrometry (HRMS), when coupled with liquid chromatography (LC–HRMS), is a powerful tool for oligonucleotide characterization. This technique enables:- Accurate molecular weight determination
- Identification of sequence variants
- Detection and localization of chemical modifications
Nuclear Magnetic Resonance (NMR) Spectroscopy
Nuclear Magnetic Resonance (NMR) spectroscopy is a valuable complementary technique for the structural characterization of oligonucleotides and their impurities. Unlike mass spectrometric methods, NMR provides direct molecular-level structural information, making it particularly useful for confirming backbone chemistry and chemical modifications.Detection of Phosphodiester (PO) Impurities
In phosphorothioate (PS)-modified oligonucleotides, phosphodiester (PO) impurities can arise due to incomplete sulfurization during synthesis. These impurities are critical to monitor because they can affect biological stability and overall drug performance. ³¹P NMR is especially well suited for detecting PO impurities because phosphorus atoms in different chemical environments produce distinct signals. Using ³¹P NMR, analysts can:- Differentiate PO and PS linkages based on their chemical shifts
- Quantify PO impurity levels through signal integration
- Observe PS stereochemistry (Rp/Sp diastereomers), which appear as characteristic split peaks
- Assess overall backbone composition without prior separation
- Confirm phosphorothioate linkage integrity
- Identify sugar and base modifications
- Evaluate conformational and structural features
Importance of High-Purity Analytical Standards
Reliable impurity analysis depends on the availability of well-characterized analytical standards. These standards are essential for:- Confirming impurity identity
- Validating analytical methods
- Enabling accurate quantification