Forced degradation studies are a critical component of pharmaceutical development for GLP-1 peptide drugs such as Liraglutide, Semaglutide, and Tirzepatide. Unlike real-time stability testing, forced degradation deliberately subjects the drug substance and drug product to extreme conditions to accelerate degradation and reveal potential impurity pathways.
Regulatory agencies rely on these studies to evaluate molecular stability, validate analytical methods, and ensure that all clinically relevant impurities have been identified and controlled.
Regulatory Expectations for Forced Degradation Studies
Regulatory agencies expect forced degradation studies to be scientifically justified, comprehensive, and product-specific, rather than generic stress testing exercises.
Key expectations include:
- Degradation pathways relevant to real storage and handling conditions
- Sufficient degradation (typically 5–20%) without complete destruction of the molecule
- Identification and characterization of major degradation products
- Clear linkage between forced degradation results and stability-indicating methods
Forced degradation data directly supports impurity control strategies reviewed during ANDA and NDA submissions. Certified GLP-1 impurity standards enable simpler and more reliable identification and characterization of degradation products.
Common Stress Conditions Applied to GLP-1 Peptide Drugs
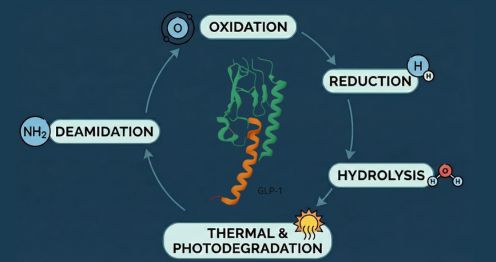
Due to their peptide backbone and sensitive amino acid residues, GLP-1 drugs are subjected to multiple orthogonal stress conditions.
- Hydrolytic Stress: Exposure to acidic, and alkaline conditions accelerates peptide bond cleavage. Hydrolysis often results in truncated peptides and fragment impurities, particularly at labile peptide bonds.
- Oxidative Stress: Oxidative conditions (e.g., hydrogen peroxide exposure) target residues such as tryptophan, tyrosine, methionine, and histidine. Oxidation leads to impurities like Trp(O), kynurenine, and related species, which are commonly observed in GLP-1 peptides.
- Thermal Stress: Elevated temperatures accelerate both hydrolysis and oxidation, often revealing aggregation tendencies and cyclic degradation products.
- Photolytic Stress: Light exposure induces photodegradation, particularly affecting aromatic amino acids. This stress is critical for injectable formulations exposed to ambient light during handling.
- Solid-State Stress (for Drug Substance): Moisture, heat, and light stress applied to solid peptide material help assess degradation risks during manufacturing, storage, and transport.
Impurity Generation and Identification Through Forced Degradation
Forced degradation enables the intentional generation of degradation-related impurities that may not be observed in early stability studies. These impurities are then:
- Separated using high-resolution HPLC or UPLC
- Identified by accurate mass measurement via HRMS
- Structurally confirmed through amino acid sequencing by HRMS, NMR, or complementary techniques
This process ensures that degradation products observed during long-term stability are already understood, characterized, and controlled.
Role of Forced Degradation in Stability-Indicating Method Validation
One of the most critical outcomes of forced degradation studies is the demonstration that analytical methods are stability-indicating.
A valid stability-indicating method must:
- Resolve the main peptide from all degradation products
- Detect impurities at low regulatory thresholds
- Accurately quantify impurities even in stressed samples
Forced degradation samples are therefore essential during method development and validation for GLP-1 peptide drugs.
Linking Forced Degradation to Lifecycle Impurity Control
Stability studies are particularly important for immunogenicity evaluation, as certain impurities may not be present at release but can emerge over shelf life. Oxidative species, deamidated variants, and cyclic degradation products often increase under accelerated or stressed conditions.
Monitoring impurity evolution during stability testing helps ensure that immunogenic risk does not increase over time, especially for long-term therapies like GLP-1 analogs.
Data generated from forced degradation studies informs:
- Selection of stability-indicating impurities
- Justification of impurity limits
- Identification of impurities requiring reference standards
- Long-term stability monitoring strategies
This linkage ensures that impurity control remains proactive rather than reactive throughout the product lifecycle.
Importance of Reference Standards in Forced Degradation Studies
While forced degradation generates impurities, certified impurity reference standards are essential for accurate quantification and regulatory defensibility. These standards allow:
- Confirmation of impurity identity
- Validation of analytical methods
- Reliable comparison with RLD impurity profiles
- Consistent monitoring across development stages
Without certified standards, forced degradation data remains qualitative and insufficient for regulatory decision-making.
Daicel Pharma Standards’ Role
Daicel Pharma Standards provides a wide range of fully characterized GLP-1 peptide impurity standards, including degradation-related impurities commonly generated during forced degradation studies. These standards support pharmaceutical companies in translating forced degradation data into robust analytical control strategies and regulator-ready submissions.
Conclusion
Forced degradation studies are not merely a regulatory checkbox for GLP-1 peptide drugs, they are foundational to understanding molecular stability, impurity formation, and long-term product safety. When combined with advanced analytical methods and certified impurity standards, forced degradation enables a scientifically sound and regulator-aligned approach to GLP-1 peptide development.
Write a comment
Your email address will not be published. All fields are required